
The Glass Transition
 We have been producing glass for more than 5000 years. Examples include tools to hunt, packaging to preserve our food, transparent panels to isolate us from cold, and thin layers where the simple touch of our fingers is transformed into information. All of these are glass. Under this generic name, we identify a class of materials showing a solid-like response, though lacking –like liquids– the long-range order of crystals.
We have been producing glass for more than 5000 years. Examples include tools to hunt, packaging to preserve our food, transparent panels to isolate us from cold, and thin layers where the simple touch of our fingers is transformed into information. All of these are glass. Under this generic name, we identify a class of materials showing a solid-like response, though lacking –like liquids– the long-range order of crystals.
Glasses are prepared by quickly cooling or pressurizing a liquid, which induces a slow-down in molecular motion. When these operations are performed at a constant rate, the timescale of structural relaxation eventually surpasses the time we are allowing for equilibration (~ the inverse of the cooling/pressuring rate). The result is that the system falls out of equilibrium. This is referred to as the glass transition.
Despite what is claimed in the introductions of a very large number of papers and doctoral theses, what occurs at the glass transition is far from being a mystery. The more crucial question is: what kind of changes at the molecular level induce such a tremendous increase in relaxation time in a liquid as it approaches the glass transition?
A large community of researchers, spread over disciplines including physics, chemistry, materials science, engineering and biology, has been seeking to provide a unified theory of the glass transition for nearly one hundred years. Despite efforts, no consensus has been reached to date on why the viscosity of liquids that cannot crystallize increases by almost 8 orders of magnitude within a limited temperature range, where density varies by not more than 1%.
Because of their complex macromolecular architecture, polymers do not crystallize easily. Polymers are thus commonly present in the form of glasses, which explains the huge interest both at the fundamental and applied level on their glass transition.
For about three decades, an impressive proportion of research on the glass transition of polymers has been dedicated to the investigation of chains confined at the nanoscale level, in geometries ranging from films (1D confinement), to pores (2D), to spheres (3D). Different theoretical approaches have indicated the presence of a temperature-dependent length scale of vitrification, ξ, which increases upon cooling following a trend characteristic of critical phenomena. Theoretical predictions indicate that this length scale reaches values on the order of 1–5 nm at the glass transition temperature. It is commonly accepted that interfacial interactions interfere with finite-size effects at length scales much larger than ξ. These interferences limit the unambiguous determination of ξ.
Nevertheless, the investigation of polymers and small molecules in confined geometries has opened an extremely rich set of new unexpected phenomena, such as adsorbed layers where molecules move faster than in unconstrained conditions, or free standing membranes where a drop in glass transition temperature as large as 70 K is accompanied by an unexpected increase in elastic modulus, or even spatial distributions of molecular relaxation times that extend over 14 macromolecular sizes. 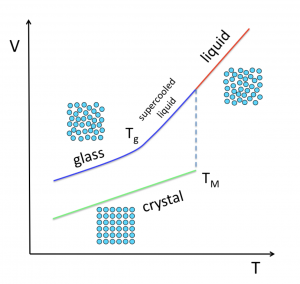
Considering the availability of existing (and excellent) reviews on the glass transition, it is worth defining here the scope of this work and its relationship to the others. In their Review published by Review on Progress in Physics (ROPP) SN, Emmanouil Glynos and Nicholas B. Tito, first address those new to the field of the polymer glass transition by providing an introduction to the phenomena of vitrification, and to the current methods used to investigate glassy dynamics. Next addressing experts in the field, they develop a picture of the glass transition based on current models and their experimental validity, and on reproducible experimental observations not yet explained by theory. We hope that their review will stimulate further discussion in the community, with the goal of motivating new research that addresses the latter cases.
The review is organized as follows. After a quick introduction to the structure and dynamics of polymers, the introductory reader is guided through the phenomenology and salient theories of the glass transition (1). The authors then focus on operational definitions of the glass transition temperature in experiments and in silico, on the link between vitrification and slower dynamics, and on the breakdown of this fundamental relation (2). In section (3) they discuss the factors affecting the glass transition; here they treat the vitrification of non-linear polymers (dendrimers, rings, stars, …), nanoscopic confinement, interfacial interactions, local order, free volume, and irreversible adsorption. SN, E. Glynos and N. B. Tito then seek new time and length scales of the glass transition (4), going beyond the search for ξ, by examining work on spatial distributions of molecular quantities in glasses. The review is concludes with a perspective (5) on future work.
The image on the upper left corner was prepared by NB Tito
For further reading:
Glass transition of polymers in bulk, confined geometries, and near interfaces